

Frequently asked questions (FAQ) and examples
Please also check the mailing list archives and the release notes.
- I want to build a model of a chimeric protein based on two known structures. Alternatively, I want to build a multi-domain protein model using templates corresponding only to the individual domains.
- I don't want to use one region of a template for construction of my model.
- I want to explicitly force certain Pro residues to the cis ω conformation.
- How can I select/remove/add a set of restraints?
- I want to change the default optimization or refinement protocol.
- I want to build an all hydrogen atom model with water molecules and other non-protein atoms (atoms in the HETATM records in the PDB file).
- How do I build a model with water molecules or residues that do not have an entry in the topology and/or parameter files?
- How do I define my own residue types, such as D-amino acids, special ligands, and unnatural amino-acids?
- How do I define my own patching residue types?
- Is it possible to restrain secondary structure in the target sequence?
- I want to patch the N-terminal or (C-terminal) residue (e.g., to model acetylation properly), but the Model.patch() command does not work.
- Is it possible to use templates with the coordinates for Cα atoms only?
- How do I analyze the output log file?
- How do I prevent “knots” in the final models?
- What is considered to be the minimum length of a sequence motif necessary to derive meaningful constraints from the alignment to use in modeling.. one, two, three, or more?
- Does Modeller have a graphical interface (GUI) ?
- What do the ‘Alignment sequence not found in PDB file’ or ‘Number of residues in the alignment and pdb files are different’ errors mean?
- Can I make a web interface or GUI for Modeller?
- I get warnings such as 'Could not find platform independent libraries', 'import site failed' or 'No module named socket'
- I want to build a model of a chimeric protein based on two
known structures. Alternatively, I want to build a multi-domain protein
model using templates corresponding only to the individual domains.
This can be accomplished using the standard AutoModel class (see Chapter 2). The alignment should be as follows when the chimera is a combination of proteins A and B:
proteinA aaaaaaaaaaaaaaaaaaaaaaaaaaaa---------------------------------- proteinB ----------------------------bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb chimera aaaaaaaaaaaaaaaaaaaaaaaaaaaabbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb
In the PIR format the alignment file is:
>P1;proteinA structureX:proteinA aaaaaaaaaaaaaaaaaaaaaaaaaaaa----------------------------------* >P1;proteinB structureX:proteinB ----------------------------bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb* >P1;chimera sequence:chimera aaaaaaaaaaaaaaaaaaaaaaaaaaaabbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb*
If no additional information is available about the relative orientation of the two domains the resulting model will probably have an incorrect relative orientation of the two domains when the overlap between A and B is non-existing or short. To obtain satisfactory relative orientation of modeled domains in such cases, orient the two template structures appropriately before the modeling.
- I don't want to use one region of a template for construction
of my model.
The easiest way to achieve this is to not align that region of the template with the target sequence. If region 'bbbbbbbb' of the template should not be used as a template for region 'eeeee' of the target sequence the alignment should be like this:
template aaaaaaaaaaaaaaaaaaaaaaaa-----bbbbbbbbcccccccccccccccccccccccccccccc target ddddddddddddddddddddddddeeeee--------ffffffffffffffffffffffffffffff
The effect of this alignment is that no homology-derived restraints will be produced for region 'eeeee'.
- I want to explicitly force certain Pro residues to the
cis ω conformation.
MODELLER should usually be allowed to handle this automatically via the omega dihedral angle restraints, which are calculated by default.
from modeller import * from modeller.automodel import * from modeller.scripts import cispeptide # Redefine the special_restraints routine to force Pro to cis conformation: # (this routine is empty by default): class MyModel(AutoModel): def special_restraints(self, aln): a = self.atoms cispeptide(self.restraints, atom_ids1=(a['O:4'], a['C:4'], a['N:5'], a['CA:5']), atom_ids2=(a['CA:4'], a['C:4'], a['N:5'], a['CA:5'])) # This is as usual: log.verbose() env = Environ() a = MyModel(env, alnfile='align1.ali', knowns='templ1', sequence='targ1') a.make() - How can I select/remove/add a set of restraints?
Restraints can be read from a file by Restraints.append(), calculated by Restraints.make() or Restraints.make_distance(), or added “manually” by Restraints.add(). Restraints.pick() picks those restraints for objective function calculation that restrain the selected atoms only. The 'AutoModel.homcsr()' routine contains examples of selecting atoms when generating restraints by Restraints.make_distance(). There are also commands for adding and unselecting single restraints, Restraints.add() and Restraints.unpick(), respectively. If you do Restraints.condense(), the unselected restraints will be deleted. This is useful for getting rid of the unwanted restraints completely.
- I want to change the default optimization or refinement protocol.
See Section 2.2.2.
- I want to build an all hydrogen atom model with water molecules and
other non-protein atoms (atoms in the HETATM records in the PDB file).
See Sections 2.2.1 and 2.2.5 for some examples.
from modeller import * from modeller.automodel import * log.verbose() env = Environ() env.io.hydrogen = env.io.hetatm = env.io.water = True a = AllHModel(env, alnfile='align1.ali', knowns='templ1', sequence='targ1') a.make()
- How do I build a model with water molecules or residues that
do not have an entry in the topology and/or parameter files?
See Section 2.2.1 for an example.
- How do I define my own residue types, such as D-amino acids,
special ligands, and unnatural amino-acids?
This is a painful area in all molecular modeling programs. However, CHARMM and X-PLOR provide a reasonably straightforward solution via the residue topology and parameter libraries. MODELLER uses CHARMM topology and parameter library format and also extends the options by allowing for a generic “BLK” residue type (Section 5.2.1). This BLK residue type circumvents the need for editing any library files, but it is not always possible to use it. Due to its conformational rigidity, it is also not as accurate as a normal residue type. In order to define a new residue type in the MODELLER libraries, you have to follow the series of steps described below. As an example, we will define the ALA residue without any hydrogen atoms. You can add an entry to the MODELLER topology or parameter file; you can also use your own topology or parameter files. For more information, please see the CHARMM manual.
- Define the new residue entry in the residue topology file (RTF),
say 'top_heav.lib'.
RESI ALA 0.00000 ATOM N NH1 -0.29792 ATOM CA CT1 0.09563 ATOM CB CT3 -0.17115 ATOM C C 0.69672 ATOM O O -0.32328 BOND CB CA N CA O C C CA C +N IMPR C CA +N O CA N C CB IC -C N CA C 1.3551 126.4900 180.0000 114.4400 1.5390 IC N CA C +N 1.4592 114.4400 180.0000 116.8400 1.3558 IC +N CA *C O 1.3558 116.8400 180.0000 122.5200 1.2297 IC CA C +N +CA 1.5390 116.8400 180.0000 126.7700 1.4613 IC N C *CA CB 1.4592 114.4400 123.2300 111.0900 1.5461 IC N CA C O 1.4300 107.0000 0.0000 122.5200 1.2297 PATC FIRS NTER LAST CTER
You can obtain an initial approximation to this entry by defining the new residue type using the residue type editor in QUANTA and then writing it to a file.
The RESI record specifies the CHARMM residue name, which can be up to four characters long and is usually the same as the PDB residue name (exceptions are the potentially charged residues where the different charge states correspond to different CHARMM residue types). The number gives the total residue charge.
The ATOM records specify the IUPAC (i.e., PDB) atom names and the CHARMM atom types for all the atoms in the residue. The number at the end of each ATOM record gives the partial atomic charge.
The BOND records specify all the covalent bonds between the atoms in the residue (e.g., there are bonds CB–CA, N–CA, O–C, etc.). In addition, symbol '+' is used to indicate the bonds to the subsequent residue in the chain (e.g., C – +N). The covalent angles and dihedral angles are calculated automatically from the list of chemical bonds.
The IMPR records specify the improper dihedral angles, generally used to restrain the planarity of various groups (e.g., peptide bonds and sidechain rings). See also below.
The IC (internal coordinate) records are used for constructing the initial Cartesian coordinates of a residue. An entry
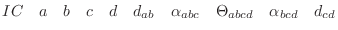
specifies distances
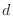 , angles α, and either dihedral angles
or improper dihedral angles Θ between atoms
, angles α, and either dihedral angles
or improper dihedral angles Θ between atoms 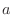 ,
, 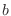 ,
, 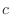 and
, given by their IUPAC names. The improper dihedral angle
is specified when the third atom, , is preceded by a star,
'*'. As before, the '-' and '+' prefixes for the atom names select
the corresponding atom from the preceding and subsequent residues,
respectively. The distances are in angstroms, angles in degrees.
The distinction between the dihedral angles and
improper dihedral angles is unfortunate since they are the
same mathematically, except that by convention when using the
equations, the order of the atoms for a dihedral angle is
and
, given by their IUPAC names. The improper dihedral angle
is specified when the third atom, , is preceded by a star,
'*'. As before, the '-' and '+' prefixes for the atom names select
the corresponding atom from the preceding and subsequent residues,
respectively. The distances are in angstroms, angles in degrees.
The distinction between the dihedral angles and
improper dihedral angles is unfortunate since they are the
same mathematically, except that by convention when using the
equations, the order of the atoms for a dihedral angle is 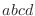 and for an improper dihedral angle it is
and for an improper dihedral angle it is 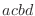 .
.
The PATC record specifies the default patching residue type when the current residue type is the first or the last residue in a chain.
- You have to make sure that all the CHARMM atom types of the
new residue type occur in the MASS records at the beginning
of the topology library: Add your entry at the end of the MASS list if
necessary. If you added any new CHARMM atom types, you also have to
add them to the radii library, 'modlib/radii.lib'
and to the solvation library, 'modlib/solv.lib'.
The radii library lists the atomic radii for the
different topology models, for the long range non-bonded
soft-sphere term. The full name of the file that is
used during calculation is given by the environment variable $RADII_LIB.
- Optionally, you can add the residue entry to the library of
MODELLER topology models, 'modlib/models.lib'. The runtime
version of this library is specified by the environment variable
$MODELS_LIB. This library specifies which subsets of atoms
in the residue are used for each of the possible topologies.
Currently, there are 10 topologies selected by Topology.submodel
(3 is default):
1 ALLH all atoms 2 POL polar hydrogens only 3 HEAV non-hydrogen atoms only 4 MCCB non-hydrogen mainchain (N, C, CA, O) and CB atoms 5 MNCH non-hydrogen mainchain atoms only 6 MCWO non-hydrogen mainchain atoms without carbonyl O 7 CA CA atoms only 8 MNSS non-hydrogen mainchain atoms and disulfide bonds 9 CA3H reduced model with a small number of sidechain interaction centers 10 CACB CA and CB atoms only The Ala entry is:
# ALLH POLH HEAV MCCB MNCH MCWO CA MNSS CA3H CACB * RESI ALA ATOM NH1 NH1 NH1 NH1 NH1 NH1 #### NH1 #### #### ATOM H HN #### #### #### #### #### #### #### #### ATOM CT1 CT1 CT1 CT1 CT1 CT1 CT1 CT1 CAH CT1 ATOM HB #### #### #### #### #### #### #### CH3E #### ATOM CT3 CT3 CT3 CT3 #### #### #### #### #### CT2 ATOM HA #### #### #### #### #### #### #### #### #### ATOM HA #### #### #### #### #### #### #### #### #### ATOM HA #### #### #### #### #### #### #### #### #### ATOM C C C C C C #### C #### #### ATOM O O O O O #### #### O #### ####The residue entries in this library are separated by stars. The '####' string indicates a missing atom. The atom names for the present atoms are arbitrary. The order of the atoms must be the same as in the CHARMM residue topology library. If a residue type does not have an entry in this library, all atoms are used for all topologies.
- You have to add the new residue type to the residue type library,
'modlib/restyp.lib'. The execution version of this file is
specified by the environment variable $RESTYP_LIB. See the comments
in the file for further information.
Every residue in the CHARMM topology file has to have an entry in the $RESTYP_LIB library, but not every residue entry in the $RESTYP_LIB library needs an entry in the residue topology file. If you need to edit the $RESTYP_LIB file, it is recommended that you change a copy of it, and provide that file to the Environ() constructor.
- In general, when you add a new residue type, you also add new chemical bonds, angles, dihedral angles, improper dihedral angles, and non-bonded interactions, new in the sense that a unique combination of CHARMM atoms types is involved whose interaction parameters are not yet specified in the parameter library (see also Section 5.2.1). In such a case, you will get a number of warning and/or error messages when you generate the stereochemical restraints by the Restraints.make() command. These messages can sometimes be ignored because MODELLER will guess the values for the missing parameters from the current Cartesian coordinates of the model. When this is not accurate enough or if the necessary coordinates are undefined you have to specify the parameters explicitly in the parameter library. Search for BOND, ANGL, DIHE, and IMPR sections in the parameters library file and use the existing entries to guess your new entries. Note that you can use dummy atom types 'X' to create general dihedral (i.e., X A A X) and improper dihedral angle (i.e., A X X A) entries, where A stands for any of the real CHARMM atom types. For the dihedral angle cosine terms, the CHARMM convention for the phase is different for 180° from MODELLER's (Eq. A.84). If you use non-bonded Lennard-Jones terms, you also have to add a NONB entry for each new atom type. If you use the default soft-sphere non-bonded restraints, you have already taken care of it by adding the new atom types to the $RADII_LIB and $RADII_LIB libraries.
- Define the new residue entry in the residue topology file (RTF),
say 'top_heav.lib'.
- How do I define my own patching residue types?
This is even messier than defining a new residue type. As an example, we will define the patching residue for establishing a disulfide bond between two CYS residues.
PRES DISU -0.36 ! Patch for disulfides. Patch must be 1-CYS and 2-CYS. ATOM 1:CB CT2 -0.10 ! ATOM 1:SG SM -0.08 ! 2:SG--2:CB-- ATOM 2:SG SM -0.08 ! / ATOM 2:CB CT2 -0.10 ! -1:CB--1:SG DELETE ATOM 1:HG DELETE ATOM 2:HG BOND 1:SG 2:SG IC 1:CA 1:CB 1:SG 2:SG 0.0000 0.0000 180.0000 0.0000 0.0000 IC 1:CB 1:SG 2:SG 2:CB 0.0000 0.0000 90.0000 0.0000 0.0000 IC 1:SG 2:SG 2:CB 2:CA 0.0000 0.0000 180.0000 0.0000 0.0000
The PRES record specifies the CHARMM patching residue name (up to four characters).
The ATOM records have the same meaning as for the RESI residue types described above. The extension is that the IUPAC atom names (listed first) must be prefixed by the index of the residue that is patched, if the patch affects multiple residues. In this example, there are two CYS residues that are patched, thus the prefixes 1 and 2. When using the Model.patch() command, the order of the patched residues specified by residues must correspond to these indices (this is only important when the patch is not symmetric, unlike the 'DISU' patch in this example).
DELETE records specify the atoms to be deleted, the two hydrogens bonded to the two sulfurs in this case.
The BOND and IC (internal coordinate) records are the same as those for the RESI residues, except that the atom names are prefixed with the patched residue indices.
- Is it possible to restrain secondary structure in the
target sequence?
Yes — see Section 2.2.11 for an example.
- I want to patch the N-terminal or (C-terminal) residue (e.g.,
to model acetylation properly), but the Model.patch() command does not work.
This is probably because the N-terminus is patched by default with the NTER patching residue (corresponding to –NH3
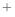 ) and a patched
residue must not be patched again. The solution is to turn the default
patching off by env.patch_default = False before the
Model.generate_topology() command is called.
) and a patched
residue must not be patched again. The solution is to turn the default
patching off by env.patch_default = False before the
Model.generate_topology() command is called.
- Is it possible to use templates with the coordinates for
Cα atoms only?
Yes. You do not have to do anything special.
- How do I analyze the output log file?
First, check for the error messages by searching for string '_E>''. These messages can only rarely be ignored. Next, check for the warning messages by searching for string '_W>''. These messages can almost always be ignored. If everything is OK so far, the most important part of the log file is the output of the Selection.energy() command for each model. This is where the violations of restraints are listed. When there are too many too violated restraints, more optimization or a different alignment is needed. What is too many and too much? It depends on the restraint type and is best learned by doing Selection.energy() on an X-ray structure or a good model to get a feel for it. You may also want to look at the output of command Alignment.check(), which should be self-explanatory. I usually ignore the other parts of the log file.
- How do I prevent “knots” in the final models?
The best way to prevent knots is to start with a starting structure that is as close to the desired final model as possible. Other than that, the only solution at this point is to calculate independently many models and hope that in some runs there won't be knots. Knots usually occur when one or more neighboring long insertions (i.e., longer than 15 residues) are modeled from scratch. The reason is that an insertion is built from a randomized distorted structure that is located approximately between the two anchoring regions. Under such conditions, it is easy for the optimizer to “fall” into a knot and then not be able to recover from it. Sometimes knots result from an incorrect alignment, especially when more than one template is used. When the alignment is correct, knots are a result of optimization not being good enough. However, making optimization more thorough by increasing the CPU time would not be worth it on the average as knots occur relatively infrequently. The excluded volume restraints are already included in standard comparative modeling with the AutoModel class (see Chapter 2).
- What is considered to be the minimum length of a sequence motif
necessary to derive meaningful constraints from the alignment to use in
modeling.. one, two, three, or more?
Usually more than that (dozens if you want just to detect reliable similarity, and even more if you want a real model). It is good to have at least 35-40% sequence identity to build a model. Sometimes even 30% is OK.
- Does Modeller have a graphical interface (GUI) ?
No; Modeller is run from the command line, and uses a Python script to direct it. Graphical interfaces to Modeller are commercially available from BIOVIA. Also, check the links page in the Modeller wiki for GUIs contributed by Modeller users.
- What do the ‘Alignment sequence not found in PDB file’ or
‘Number of residues in the alignment and pdb files are different’ errors mean?
When you give MODELLER an alignment, it also needs to read the structure of the known proteins (templates) from PDB files. In order to correctly match coordinates to the residues specified in the alignment, the sequences in the PDB file and the alignment file must be the same (although obviously you can add gap or chain break characters to your alignment). If they are not, you see this error. (Note that MODELLER takes the PDB sequence from the ATOM and HETATM PDB records, not the SEQRES records.) You should also check the header of your alignment file, to make sure that you are reading the correct chain and residue numbers from your PDB.
To see the sequence that MODELLER reads from the PDB file '1BY8.pdb', use this short script to produce a '1BY8.seq' sequence file:
from modeller import * env = Environ() # If you also want to see HETATM residues, uncomment this line: #env.io.hetatm = True code = '1BY8' mdl = Model(env, file=code) aln = Alignment(env) aln.append_model(mdl, align_codes=code) aln.write(file=code+'.seq')
- Can I make a web interface or GUI for Modeller?
Certainly, although you should bear in mind that the Modeller license is non-transferable, and permits free usage only for academic purposes.
For web interfaces, users must obtain their own Modeller license key directly from us; your web interface should provide a text box into which users should put their license key, and then use that input to set the KEY_MODELLER10v3 environment variable, as is done by our own MODWEB and MODLOOP interfaces. (Note that you will first need to edit the file modlib/modeller/config.py in your Modeller installation to remove the line that sets the license, since this takes precedence over the environment variable setting.)
For GUIs or other interfaces (e.g. frameworks), users should obtain and license Modeller directly from us, rather than it being bundled with your software.
In all cases, please update the links page in the Modeller wiki, to advertise your software to Modeller users.
- I get warnings such as 'Could not find platform independent
libraries', 'import site failed' or 'No module named socket'
These refer to missing Python modules on your system. In the first two cases, these are just warnings that can be safely ignored - most Modeller scripts do not need Python modules anyway, and will run successfully. However, some Modeller scripts, such as the parallel task support, do need modules (such as socket) and will not function without them. Please refer to the release notes for two possible solutions in this case.