

Tutorial
Advanced example:
Modeling of a protein-ligand complex based on multiple templates, loop
refinement and user specified restraints.
All input and output files for this example are available to download, in either zip format (for Windows) or .tar.gz format (for Unix/Linux).
For this example we will not describe step-by-step all MODELLER commands. Please, refer to the basic-example in the tutorial for more details.
An important aim of modeling is to contribute to understanding of the function of the modeled protein. Inspection of the 1bdm:A template structure (built in the basic modeling tutorial) revealed that loop 93-100, one of the functionally most important parts of the enzyme, is disordered and does not appear in the PDB structure. Most probably the long active site loop is flexible in the absence of a ligand and could not be seen in the diffraction map. The unreliability of the template coordinates and the inability of MODELLER to model long insertions is why this loop was poorly modeled in TvLDH, as indicated by the DOPE profile.
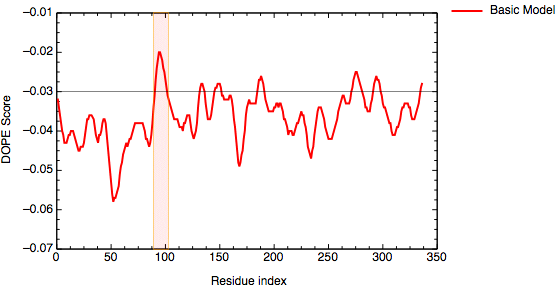
DOPE score profile for model TvLDH.B99990001
Since we are interested in understanding differences in specificity between two similar proteins, we need to build precise and accurate models. Therefore, we need to find new strategies to increase the accuracy of the models. In this example, we will explore three different approaches:
- Use of multiple templates.
- Modeling the loop using ab-initio methods.
- Modeling using a known ligand bound to the binding site.
Multiple templates
The structure of the Malate Dehydrogenase 1bdm has been clustered in the DBAli database within the family fm00495 of 4 members (2mdh:A, 2mdh:B. 1b8p:A and 1bdm:A). The multiple alignment generated by the command salign() in MODELLER is used in DBAli to generate a multiple structure alignment of the family. The alignment can be downloaded from the DBAli database or you can use the file `salign.py' to calculate it on your computer.
# Illustrates the SALIGN multiple structure/sequence alignment
from modeller import *
log.verbose()
env = Environ()
env.io.atom_files_directory = ['.', '../atom_files/']
aln = Alignment(env)
for (code, chain) in (('2mdh', 'A'), ('1bdm', 'A'), ('1b8p', 'A')):
mdl = Model(env, file=code, model_segment=('FIRST:'+chain, 'LAST:'+chain))
aln.append_model(mdl, atom_files=code, align_codes=code+chain)
for (weights, write_fit, whole) in (((1., 0., 0., 0., 1., 0.), False, True),
((1., 0.5, 1., 1., 1., 0.), False, True),
((1., 1., 1., 1., 1., 0.), True, False)):
aln.salign(rms_cutoff=3.5, normalize_pp_scores=False,
rr_file='$(LIB)/as1.sim.mat', overhang=30,
gap_penalties_1d=(-450, -50),
gap_penalties_3d=(0, 3), gap_gap_score=0, gap_residue_score=0,
dendrogram_file='fm00495.tree',
alignment_type='tree', # If 'progresive', the tree is not
# computed and all structues will be
# aligned sequentially to the first
feature_weights=weights, # For a multiple sequence alignment only
# the first feature needs to be non-zero
improve_alignment=True, fit=True, write_fit=write_fit,
write_whole_pdb=whole, output='ALIGNMENT QUALITY')
aln.write(file='fm00495.pap', alignment_format='PAP')
aln.write(file='fm00495.ali', alignment_format='PIR')
aln.salign(rms_cutoff=1.0, normalize_pp_scores=False,
rr_file='$(LIB)/as1.sim.mat', overhang=30,
gap_penalties_1d=(-450, -50), gap_penalties_3d=(0, 3),
gap_gap_score=0, gap_residue_score=0, dendrogram_file='1is3A.tree',
alignment_type='progressive', feature_weights=[0]*6,
improve_alignment=False, fit=False, write_fit=True,
write_whole_pdb=False, output='QUALITY')
File: multiple_template/salign.py
The reads in all of the sequences from PDB files (using the append_model command), and then uses salign multiple times, to generate an initial rough alignment and then improve upon it by using more information. The alignment is then written out in both PIR and PAP formats, and a quality score is calculated by calling salign one more time.
After inspecting the multiple structure alignment it is evident that chain B of 2mdh contains an unusual number of LYS residues. The HEADER of the PDB file indicates that the sequence of the protein was unknown at the time of refinement and it was difficult to identify most of the residues in the structure. Therefore, the 2mdh:B entry was removed from the multiple structure alignment.
_aln.pos 10 20 30 40 50 60 2mdhA GSMQIRVLVTG-AAQLAFTLLYSIGDGSVFGKNQPILLSLMDVVP--KQQTSEAVNMQLQNCALP-LL 1bdmA MKAPVRVAVTGAAGQIGYSLLFRIAAGEMLGKDQPVILQLLEIPQ--AMKALEGVVMELEDCAFPLLA 1b8pA -KTPMRVAVTGAAGQICYSLLFRIANGDMLGKDQPVILQLLEIPNEKAQKALQGVMMEIDDCAFPLLA _consrvd ** *** * * ** * * ** ** * * * * ** * * _aln.p 70 80 90 100 110 120 130 2mdhA KSQFGKNSGN-YASQNVGVLLAGQRAKNAAKN---LKANVKIFKCQGAALNKYWKKSVIVIVVGNPAT 1bdmA GLEATDDPDVAFKDADYALLVGAAPR---------LQVNGKIFTEQGRALAEVAKKDVKVLVVGNPAN 1b8pA GMTAHADPMTAFKDADVALLVGARPRGPGMERKDLLEANAQIFTVQGKAIDAVASRNIKVLVVGNPAN _consrvd * * * ** ** * * ****** _aln.pos 140 150 160 170 180 190 200 2mdhA NNCLTASKNSAQLNKAKQVNSVKLNHNRAKSMLSQKLGNSPKLSKNVILYGQHGQSQFSGLIQLQLQN 1bdmA TNALIAYKNAPGLNPRNFTAMTRLDHNRAKAQLAKKTGTGVDRIRRMTVWGNHSSIMFPDLFHAEVD- 1b8pA TNAYIAMKSAPSLPAKNFTAMLRLDHNRALSQIAAKTGKPVSSIEKLFVWGNHSPTMYADYRYAQID- _consrvd * * * * * **** * * * * _aln.pos 210 220 230 240 250 260 270 2mdhA KQSAGVR-ASKNQSWKTSIYNNVIQQRGVVHVQARTANNSMKTGFALNLYVKHLWKGISQ-KLAQMGL 1bdmA --GRPALELV-DMEWYEKVFIPTVAQRGAAIIQARGASSAASAANAAIEHIRDWALGTPEGDWVSMAV 1b8pA --GASVKDMINDDAWNRDTFLPTVGKRGAAIIDARGVSSAASAANAAIDHIHDWVLGTAG-KWTTMGI _consrvd * ** ** * * * _aln.pos 280 290 300 310 320 330 2mdhA IAHGKAAASPKQNFSCVTRLQNKTWKIVEGLPINDFSREKMNETAKELAEEETEFAEKNSNA 1bdmA PSQGEYGIPEGIVYSFPVTAKDGAYRVVEGLEINEFARKRMEITAQELLDEMEQVKALGLI- 1b8pA PSDGSYGIPEGVIFGFPVTTENGEYKIVQGLSIDAFSQERINVTLNELLEEQNGVQ-HLLG- _consrvd * * ** * * * ** *
File: multiple_template/fm00495.pap
As for the basic example in the tutorial, next we need to align our query sequence to the template structures. For that task we again use the salign() command (file `align2d_mult.py'). We set the align_block parameter to equal the number of structures in the template alignment, len(aln), (i.e. 3), and request a pairwise alignment, since we do not want to change the existing alignment between the templates. By setting gap_function we request the use of a structure-dependent gap penalty, using structural information for these 3 sequences. Only sequence information is used for the final TvLDH sequence.
from modeller import *
log.verbose()
env = Environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib')
# Read aligned structure(s):
aln = Alignment(env)
aln.append(file='fm00495.ali', align_codes='all')
aln_block = len(aln)
# Read aligned sequence(s):
aln.append(file='TvLDH.ali', align_codes='TvLDH')
# Structure sensitive variable gap penalty sequence-sequence alignment:
aln.salign(output='', max_gap_length=20,
gap_function=True, # to use structure-dependent gap penalty
alignment_type='PAIRWISE', align_block=aln_block,
feature_weights=(1., 0., 0., 0., 0., 0.), overhang=0,
gap_penalties_1d=(-450, 0),
gap_penalties_2d=(0.35, 1.2, 0.9, 1.2, 0.6, 8.6, 1.2, 0., 0.),
similarity_flag=True)
aln.write(file='TvLDH-mult.ali', alignment_format='PIR')
aln.write(file='TvLDH-mult.pap', alignment_format='PAP')
File: multiple_template/align2d_mult.py
Next, we build the new model for the TvLDH target sequence based on the alignment against the multiple templates using the `model_mult.py' file:
from modeller import *
from modeller.automodel import *
env = Environ()
a = AutoModel(env, alnfile='TvLDH-mult.ali',
knowns=('1bdmA','2mdhA','1b8pA'), sequence='TvLDH')
a.starting_model = 1
a.ending_model = 5
a.make()
File: multiple_template/model_mult.py
Finally, we use the DOPE potential to evaluate the new model coordinates using the `evaluate_model.py' file:
from modeller import *
from modeller.scripts import complete_pdb
log.verbose() # request verbose output
env = Environ()
env.libs.topology.read(file='$(LIB)/top_heav.lib') # read topology
env.libs.parameters.read(file='$(LIB)/par.lib') # read parameters
# read model file
mdl = complete_pdb(env, 'TvLDH.B99990001.pdb')
# Assess all atoms with DOPE:
s = Selection(mdl)
s.assess_dope(output='ENERGY_PROFILE NO_REPORT', file='TvLDH.profile',
normalize_profile=True, smoothing_window=15)
File: multiple_template/evaluate_model.py
The evaluation of the model indicates that the problematic loop (residues 90 to 100) has improved by using multiple structural templates. The global DOPE score for the models also improved from -38999.7 to -39164.4. MODELLER was able to use the variability in the loop region from the three templates to generate a more accurate conformation of the loop. However, the conformation of a loop in the region around the residue 275 at the C-terminal end of the sequence has higher DOPE score than for the model based on a single template.
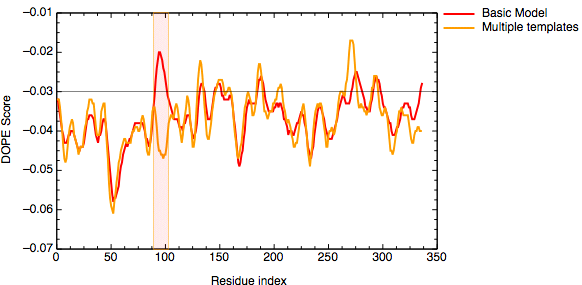
DOPE score profile for model TvLDH.B99990001.pdb
We will use the LoopModel class in MODELLER to refine the conformation of the loop between residues 273 and 283 (in the A chain). We will use the model number 1 created in the previous example as a starting structure to refine the loop. You can find this structure renamed as `TvLDH-mult.pdb' in the loop_modeling subdirectory.
Loop refining
The loop optimization method relies on a scoring function and optimization schedule adapted for loop modeling. It is used automatically to refine comparative models if you use the LoopModel class rather than AutoModel; see the example below.
# Loop refinement of an existing model
from modeller import *
from modeller.automodel import *
log.verbose()
env = Environ()
# directories for input atom files
env.io.atom_files_directory = ['.', '../atom_files']
# Create a new class based on 'LoopModel' so that we can redefine
# select_loop_atoms (necessary)
class MyLoop(LoopModel):
# This routine picks the residues to be refined by loop modeling
def select_loop_atoms(self):
# 10 residue insertion
return Selection(self.residue_range('273:A', '283:A'))
m = MyLoop(env,
inimodel='TvLDH-mult.pdb', # initial model of the target
sequence='TvLDH') # code of the target
m.loop.starting_model= 1 # index of the first loop model
m.loop.ending_model = 10 # index of the last loop model
m.loop.md_level = refine.very_fast # loop refinement method; this yields
# models quickly but of low quality;
# use refine.slow for better models
m.make()
File: loop_modeling/loop_refine.py
In this example, the LoopModel class is used to refine a region of an existing coordinate file. Note that this example also redefines the LoopModel.select_loop_atoms routine. This is necessary in this case, as the default selection selects all gaps in the alignment for refinement, and in this case no alignment is available. You can still redefine the routine, even if you do have an alignment, if you want to select a different region for optimization. Note that for the sake of time, we will be building only 10 different independently optimized loop conformations by setting the loop.ending_model parameter to 10. The next image shows the superimposition of the 10 conformations of the loop modeling. In blue, green and red we have marked the initial, best and worst loop conformations as scored by DOPE, respectively.
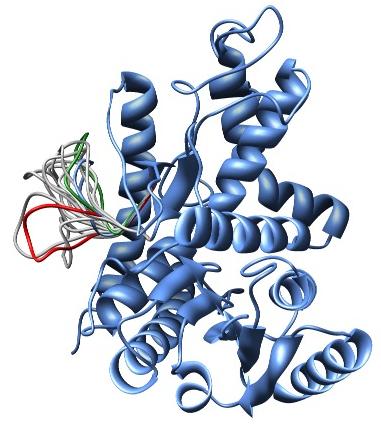
Superimposition of all 10 calculated loop conformations rendered by Chimera.
The file `model_energies.py' computes the DOPE score for all built models by using a Python for loop. The best energy loop corresponds to the 8th model (file: `model_energies.py') with a global DOPE score of -39099.1. Its energy profile calculated by `evaluate_model.py' is shown next.
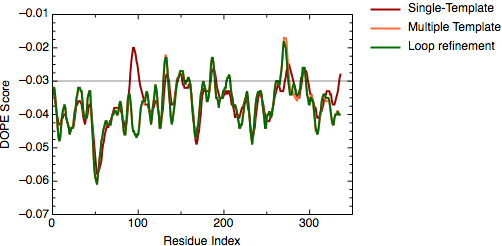
DOPE score profile for model TvLDH.BL00080001.pdb
There is only a very small increase of global DOPE score by ab-initio refinement of the loop. However, there is a small decrease in the DOPE score in the region of the loop. Therefore, we will continue the next step using the best refined structure (file: `TvLDH.BL00080001.pdb'), which is renamed in the ligand directory as `TvLDH-loop.pdb'. It is important to note that a most accurate approach to loop refinement requires the modeling of hundreds of independent conformations and their clustering to select the most representative structures of the loop.
Modeling ligands in the binding site
1emd, a malate dehydrogenase from E. coli, was identified in PDB. While the 1emd sequence shares only 32% sequence identity with TvLDH, the active site loop and its environment are more conserved. The loop for residues 90 to 100 in the 1emd structure is well resolved. Moreover, 1emd was solved in the presence of a citrate substrate analog and the NADH cofactor. The new alignment in the PAP format is shown below (file `TvLDH-1emd_bs.pap').
_aln.pos 10 20 30 40 50 60 TvLDH MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLA TvLDH_model MSEAAHVLITGAAGQIGYILSHWIASGELYGDRQVYLHLLDIPPAMNRLTALTMELEDCAFPHLA 1emd ----------------------------------------------------------------- _consrvd _aln.pos 70 80 90 100 110 120 130 TvLDH GFVATTDPKAAFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGN TvLDH_model GFVATTDPKAAFKDIDCAFLVASMPLKPGQVRADLISSNSVIFKNTGEYLSKWAKPSVKVLVIGN 1emd ----------------------GVRRKPGMDRSDLFNVN-------------------------- _consrvd *** * ** * _aln.pos 140 150 160 170 180 190 TvLDH PDNTNCEIAMLHAKNLKPENFSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLT TvLDH_model PDNTNCEIAMLHAKNLKPENFSSLSMLDQNRAYYEVASKLGVDVKDVHDIIVWGNHGESMVADLT 1emd ----------------------------------------------------------------- _consrvd _aln.pos 200 210 220 230 240 250 260 TvLDH QATFTKEGKTQKVVDVLDHDYVFDTFFKKIGHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTA TvLDH_model QATFTKEGKTQKVVDVLDHDYVFDTFFKKIGHRAWDILEHRGFTSAASPTKAAIQHMKAWLFGTA 1emd ----------------------------------------------------------------- _consrvd _aln.pos 270 280 290 300 310 320 TvLDH PGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVVEGFKVNDWLREKLDFTEKDLFHEKE TvLDH_model PGEVLSMGIPVPEGNPYGIKPGVVFSFPCNVDKEGKIHVVEGFKVNDWLREKLDFTEKDLFHEKE 1emd ----------------------------------------------------------------- _consrvd _aln.pos 330 TvLDH IALNHLAQGG/.. TvLDH_model IALNHLAQGG/-- 1emd ----------/.. _consrvd
File: ligand/TvLDH-1emd_bs.pap
The modified alignment refers to an edited 1emd structure (1emd_bs), as a second template. The alignment corresponds to a model that is based on 1emd_bs in its active site loop and on TvLDH_model, which corresponds to the best model from the previous step, in the rest of the fold. Four residues on both sides of the active site loop are aligned with both templates to ensure that the loop has a good orientation relative to the rest of the model.
The modeling script below has several changes with respect to `model-single.py'. First, the name of the alignment file assigned to alnfile is updated. Next, the variable knowns is redefined to include both templates. Another change is an addition of the `env.io.hetatm = True' command to allow reading of the non-standard pyruvate and NADH residues from the input PDB files. The script is shown next (file `model-multiple-hetero.py').
from modeller import *
from modeller.automodel import *
class MyModel(AutoModel):
def special_restraints(self, aln):
rsr = self.restraints
for ids in (('NH1:161:A', 'O1A:336:B'),
('NH2:161:A', 'O1B:336:B'),
('NE2:186:A', 'O2:336:B')):
atoms = [self.atoms[i] for i in ids]
rsr.add(forms.UpperBound(group=physical.upper_distance,
feature=features.Distance(*atoms),
mean=3.5, stdev=0.1))
env = Environ()
env.io.hetatm = True
a = MyModel(env, alnfile='TvLDH-1emd_bs.ali',
knowns=('TvLDH_model','1emd'), sequence='TvLDH')
a.starting_model = 1
a.ending_model = 5
a.make()
File: ligand/model-multiple-hetero.py
A ligand can be included in a model in two ways by MODELLER. The first case corresponds to the ligand that is not present in the template structure, but is defined in the MODELLER residue topology library. Such ligands include water molecules, metal ions, nucleotides, heme groups, and many other ligands (see question 8 in the the MODELLER FAQ). This situation is not explored further here. The second case corresponds to the ligand that is already present in the template structure. We can assume either that the ligand interacts similarly with the target and the template, in which case we can rely on MODELLER to extract and satisfy distance restraints automatically, or that the relative orientation is not necessarily conserved, in which case the user needs to supply restraints on the relative orientation of the ligand and the target (the conformation of the ligand is assumed to be rigid). The two cases are illustrated by the NADH cofactor and pyruvate modeling, respectively. Both NADH and cofactor are indicated by the `.' characters at the end of each sequence in the alignment file above (the `/' character indicates a chain break). In general, the `.' character in MODELLER indicates an arbitrary generic residue called a ``block'' residue (for details see the section on block residues in the MODELLER manual). Note that the `.' characters are present both in one of the template structures and in the model sequence. The former tells MODELLER to read the ligands from the template, and the latter tells it to include the ligands in the model. The 1emd structure file contains a citrate substrate analog. To obtain a model with pyruvate, the physiological substrate of TvLDH, we convert the citrate analog in 1emd into pyruvate by deleting the group CH(COOH)2, thus obtaining the 1emd_bs template file. A major advantage of using the `.' characters is that it is not necessary to define the residue topology.
To obtain the restraints on pyruvate, we first superpose the structures of several LDH and MDH enzymes solved with ligands. Such a comparison allows us to identify absolutely conserved electrostatic interactions involving catalytic residues Arg161 and His186 on one hand, and the oxo groups of the lactate and malate ligands on the other hand. The modeling script can now be expanded by creating a new class 'MyModel', which is derived from AutoModel but which differs in one important respect: the special_restraints routine is redefined to add, to the default restraints, some user defined distance restraints between the conserved atoms of the active site residues and their substrate. In this case, a harmonic upper bound restraint of 3.5±0.1Å is imposed on the distances between the three specified pairs of atoms. A trick is used to prevent MODELLER from automatically calculating distance restraints on the pyruvate-TvLDH complex; the ligand in the 1emd_bs template is moved beyond the upper bound on the ligand-protein distance restraints (i.e., 10).
The final selected model (shown in the ribbons image below) has a DOPE global score of -37640.9. The DOPE score is increased due to the new interactions of the protein with the ligand that are not accounted when calculating the DOPE score.
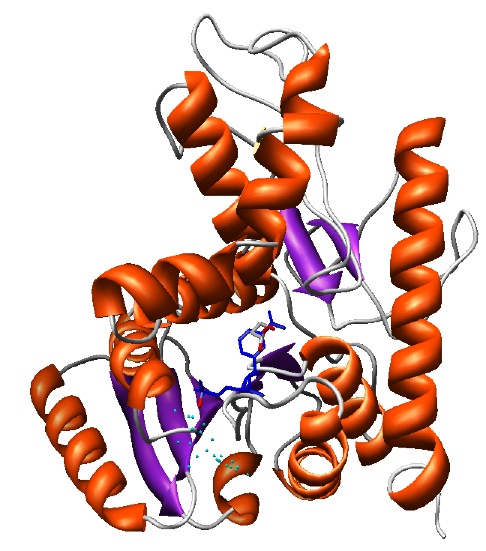
Final model with NAD and LAC ligands in the binding site rendered by Chimera.